



产品货号:
YT027
中文名称:
基因定点突变试剂盒
英文名称:
Site-directed Gene Mutagenesis Kit
产品规格:
10T|50T
发货周期:
1~3天
产品价格:
询价
简要说明:
工作原理:
本试剂盒利用了目前最常用的基因点突变技术,只需通过基于PCR的突变质粒的生成,和基于DpnI的模板质粒的消化,转化培养以及后续的酶切或测序鉴定,即可得到预期的突变质粒(参考图1)。DpnI消化仅需5分钟,累计操作时间不足2小时即可完成基因的定点突变。
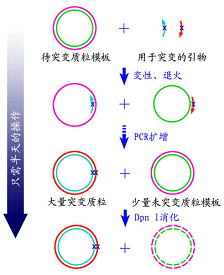
图1.基因定点突变试剂盒原理图。使用本试剂盒时需要先设计长度通常为25~45个碱基互补的两个引物,在引物中含有预期的突变位点。然后以待突变的质粒为模板,用这两个引物进行PCR扩增反应。这样可以产生含有预期的突变位点的双链质粒,但这个双链质粒中有两个nick位点。待突变的质粒通常来源于DH5α等dam+大肠杆菌,在这些dam+细菌中质粒会被甲基化修饰,而在体外通过PCR扩增得到的质粒不会被甲基化。这样用特异性识别甲基化位点的酶DpnI处理,可以消化掉待突变的质粒模板,而使通过PCR扩增出来的含有突变位点的质粒被选择性地保留下来。随后把DpnI处理过的产物转化感受态细菌后,突变质粒中的两个nick位点可以被大肠杆菌修复,得到的克隆就会含有预期的突变质粒了。
应用效果:
本试剂盒分别使用6kb和12kb的质粒进行了点突变测试,实测突变率接近100%(参考图2)。实际进行点突变时,可能会因为突变引物的设计、模板使用量、感受态效率等因素出现不同的突变率。

图2.本试剂盒实际使用效果图。
产品组成:
保存:-20℃,有效期一年。
注意事项:
使用方法:
常见问题:
相关搜索:基因定点突变试剂盒,定点突变试剂盒,Site-directed Gene Mutagenesis Kit
本试剂盒可以用于点突变,多个邻近密码子的突变,单个或多个邻近密码子的缺失或插入。基因定点突变常用于研究基因的转录调控、RNA或蛋白质的结构和功能,以及用于质粒中部分序列的微调等。本试剂盒提供了DH5α甘油菌,可用于感受态细菌的制备。
本试剂盒使用了最新的保真性更强,灵敏度更高,扩增速度可达2kb/min,扩增长度可达12kb的BalbFusion DNA Polymerase,从而大大缩短了突变反应所需的时间,进一步提高了基因定点突变的成功率。同时进一步严格验证了试剂盒中的DpnI,确保能在5分钟内充分消化掉甲基化的模板质粒,同时不会消化未甲基化的PCR产物。
工作原理:
本试剂盒利用了目前最常用的基因点突变技术,只需通过基于PCR的突变质粒的生成,和基于DpnI的模板质粒的消化,转化培养以及后续的酶切或测序鉴定,即可得到预期的突变质粒(参考图1)。DpnI消化仅需5分钟,累计操作时间不足2小时即可完成基因的定点突变。
图1.基因定点突变试剂盒原理图。使用本试剂盒时需要先设计长度通常为25~45个碱基互补的两个引物,在引物中含有预期的突变位点。然后以待突变的质粒为模板,用这两个引物进行PCR扩增反应。这样可以产生含有预期的突变位点的双链质粒,但这个双链质粒中有两个nick位点。待突变的质粒通常来源于DH5α等dam+大肠杆菌,在这些dam+细菌中质粒会被甲基化修饰,而在体外通过PCR扩增得到的质粒不会被甲基化。这样用特异性识别甲基化位点的酶DpnI处理,可以消化掉待突变的质粒模板,而使通过PCR扩增出来的含有突变位点的质粒被选择性地保留下来。随后把DpnI处理过的产物转化感受态细菌后,突变质粒中的两个nick位点可以被大肠杆菌修复,得到的克隆就会含有预期的突变质粒了。
应用效果:
本试剂盒分别使用6kb和12kb的质粒进行了点突变测试,实测突变率接近100%(参考图2)。实际进行点突变时,可能会因为突变引物的设计、模板使用量、感受态效率等因素出现不同的突变率。
图2.本试剂盒实际使用效果图。
- 使用本试剂盒进行点突变获得的LB平板照片。以一个6kb质粒为模板,使用本试剂盒进行PCR扩增以实现点突变,DpnI 37℃消化5min,取10μL产物转化100μL DH5α感受态,涂板后37℃培养过夜所获得的LB平板照片。
- 阴性对照LB平板照片。与A相比没有添加BalbFusion DNA Polymerase,这样模板质粒完全被DpnI酶所消化,没有任何的假阳性克隆产生。
- 突变前后的测序图。突变前的测序结果(WT,wild type)和从图A中挑取9个克隆的测序结果(Mu,mutant)。箭头所示为拟突变的位点,红色下划线的为突变前和突变后的碱基。本次实验的突变率达到了100%。
产品组成:
| 组分 | 10T | 50T |
| BalbFusion DNA Polymerase | 10μL | 50μL |
| 10×BalbFusion Buffer (with Mg2+) | 100μL | 500μL |
| dNTP Mix (2.5mM each) | 100μL | 500μL |
| DpnI | 10μL | 50μL |
| DH5α甘油菌 | 200μL | 1mL |
| Nuclease-Free Water | 1mL | 5mL |
保存:-20℃,有效期一年。
注意事项:
- 需自行配制LB液体培养基和LB平板以用于细菌的培养。
- 需自行设计和合成用于基因定点突变的引物。需自备用于细菌转化的试剂。
- 使用本试剂盒前请先阅读后面的常见问题。
- 本制品仅限于专业人员的科学研究用,不得用于临床诊断或治疗,不得用于食品或药品。
- 为了您的安全和健康,请穿实验服并戴一次性手套操作。
使用方法:
- 引物设计:
用于特定基因突变的引物需要单独设计,请参考如下一些基本原则进行设计:- 共需设计两条互补的引物。可以先集中设计一条,然后就可以得到互补的另一条引物。
- 引物的长度通常为25~45个碱基。
- 利用如下公式进行引物Tm值的估算,通常Tm值应该不低于78℃。
Tm = 81.5 + 0.41(%GC) - (675/N) -%mismatch
N表示引物所含碱基总数
%GC表示引物中GC碱基数占引物总碱基数的百分值,例如GC含量为50%,那么%GC就是50。
%mismatch表示引物中突变碱基数占引物总碱基数的百分值,例如错配率是2.5%,那么%mismatch就是2.5。
例一:对于模板序列5'-CCAATTTCGAGGAATTAGAACCTTATTTTGAACTTACTGAAGG-3',其中带有灰色阴影的A为待突变位点,需要突变为C,设计正向突变引物如下:
5'-CCAATTTCGAGGAATTAGCACCTTATTTTGAACTTACTGAAGG-3'
Tm = 81.5 + 0.41*(15/43)*100 - (675/43) - (1/43)*100 = 77.8
例二:对于模板序列5'-GGGAGCTCACCAAGCTGAAGAGCACCTACATTGA-3',其中两个有灰色阴影的A为待突变位点,均需要突变为G,设计正向突变引物如下:
5'-GGGAGCTCACCAGGCTGAGGAGCACCTACATTGA-3'
Tm = 81.5 + 0.41*(20/34)*100 - (675/34) - (2/34)*100 = 79.9
对于插入、缺失型的突变引物,推荐利用如下公式进行引物Tm值的估算:
Tm=81.5 + 0.41(%GC)?(675/N)
此公式中,N不包含插入或缺失的碱基。因为如果N包含插入或缺失的碱基,数字可能会比较大,从而影响计算结果。 - 也可以利用如下公式进行引物设计。
引物中突变位点任何一侧都必需满足4*(GC碱基数) + 2*(AT碱基数) ≥ 45。但引物也不宜过长,否则通常会形成非常稳定的二级结构。通常把突变位点两侧的碱基数控制在15个左右,且使两侧按照上述计算得到的数值相近。
例如引物为AGTCAGGCCAATTCGAAGCAGTCGAATTGCCAAG,其中有灰色阴影的AAG为突变位点,则
左侧:4*(GC碱基数) + 2*(AT碱基数) = 4*8 + 2*7 = 46 ≥ 45
右侧:4*(GC碱基数) + 2*(AT碱基数) = 4*8 + 2*8 = 48 ≥ 45 - 上述c和d这两种方法的计算标准有较明显的差异,但经过多次测试,两种方法都可以顺利获得预期的突变。
- 在可能的情况下,尽量把引物的GC含量控制在40~60%。
- 在可能的情况下,尽量使引物不要产生非常稳定的二级结构和引物二聚体。二级结构和二聚体可通过一些软件进行分析。
- 最好使用经过PAGE纯化的引物或更高纯度的引物。
- 共需设计两条互补的引物。可以先集中设计一条,然后就可以得到互补的另一条引物。
- 引物的配制:
如果合成得到的一个引物A的量是20nmol,另外一个互补引物B的量是19nmol。在引物A中加入200μL水,配制成浓度为100μM,在引物B中加入190μL水也配制成浓度为100μM。吸取20μL 100μM引物A和20μL 100μM引物B到一新的离心管中,再加入160μL水,混匀后即可得到可以直接用于基因定点突变反应的引物(10μM each)。 - 待突变模板质粒的选择:
选择GC含量在40~55%的待突变模板质粒,并且每一个50bp左右的局部GC含量最好也不超过70%。如果GC含量过高,请先把目的基因克隆到其它合适的载体上,再进行基因定点突变反应。另外目的基因GC含量最好也在40~55%的范围,并且每一个50bp左右的局部GC含量最好也不超过70%。如果目的基因GC含量过高,而突变位点不在高GC含量区域,可以先把该基因的不含高GC含量的一个区域克隆出来,进行定点突变,然后再把突变后的片断克隆回原基因中。如果必需使用高GC含量的质粒模板,或者有局部高GC含量的质粒模板,请另外使用专门用于高GC含量模板的PCR反应试剂。
必须使用从dam+的大肠杆菌(这类菌中质粒可以被甲基化)中抽提得到的质粒用于基因定点突变。常用的大部分大肠杆菌都是dam+的,包括DH5α、JM109等。 - 基因定点突变反应:
- 如下设置基因定点突变反应体系:
成分 扩增片段小于6kb 扩增片段大于6kb 最终浓度 体积 最终浓度 体积 Nuclease-Free Water - ?μL - ?μL 10×BalbFusion Buffer (with Mg2+) 1X 5μL 1X 5μL 引物混合物(10μM each) 0.4μM each 2μL 0.8μM each 4μL dNTP mix (2.5mM each) 0.25mM each 5μL 0.5mM each 10μL 待突变模板质粒 200ng ?μL 200ng ?μL BalbFusion DNA Polymerase 1/50 1μL 1/50 ?1μL 总体积 - 50μL - 50μL
按照上面的顺序依次加入各种试剂。在上述反应体系中,根据待突变模板质粒的量,计算出需加入的Nuclease-Free Water的量,使总体积为49μL。适当混匀后,加入1μlBalbFusion DNA Polymerase,混匀。如果用的PCR仪没有热盖,在反应体系上加入一滴矿物油(mineral oil)以防止蒸发。 - 按照如下参数设置PCR仪:
步骤 循环数 温度 <6kb时的时间 >6kb时的时间 说明 1 1 95℃ 3min 3min 最初变性 2 20 95℃ 30sec 30sec 变性 55℃ 30sec 30sec 退火 68℃ 30sec/kb 60sec/kb 延伸 3 1 68℃ 10min 15min 延伸、补全 4 1 4℃ 长时间保持 长时间保持 暂时存放
说明:上面表格中30sec/kb表示,如果待突变的质粒为6kb,那么68℃的延伸时间为3分钟。60sec/kb的含义相同。
- 如下设置基因定点突变反应体系:
- DpnI消化:
PCR反应后,直接在PCR反应体系中加入1μL DpnI,混匀后37℃孵育5min。37℃孵育可以在PCR仪上进行,也可以在水浴锅中进行。DpnI消化完毕后可以直接用于转化,或者-20℃保存备用。 - 转化、挑克隆鉴定:
感受态细菌的转化效率必需至少在107以上,否则很难得到克隆。根据所使用的感受态细菌,加入尽量多的经过DpnI消化后的突变产物用于转化。通常每100μL感受态细菌中可以加入5~10μL经过DpnI消化后的突变产物。按照所使用的感受态细菌的操作方法进行操作,在涂板前通过离心浓缩的办法,把所有被转化后的细菌,全部涂布到含有适当抗生素的平板上,培养过夜。通常会得到100个以下的克隆。如果发现有上千个克隆,那么通常是有什么地方出了问题。
对于得到的克隆,可以挑取3~5个克隆送样测序去测序,以确认得到的克隆是否是预期的突变克隆。通常大部分的是预期的突变克隆。但有时也可能会因为随机因素,会测序了3~5个克隆才得到一个预期的突变克隆。
常见问题:
- 转化后没有克隆或克隆数极少:
- 感受态细菌效率不够高,请检测一下感受态细胞的效率,确保转化效率在107以上,当然高一些更好。
- 把DpnI消化后的产物用常规的乙醇沉淀方法进行沉淀,然后溶解在较小的体积中。这样就可以把所有的产物全部用于转化。
- 优化基因定点突变中的PCR参数。可以把最初的95℃变性时间延长为5min,循环中95℃变性的时间延长至1min,把循环中的68℃的延伸时间改为1min/kb至2min/kb,退火可以改为60~55℃或65~55℃等的touch down,退火时间也可以适当延长。
- 引物设计有问题。通过突变反应中的PCR没有很好地扩增出预期的突变质粒。
- 如果使用矿物油(mineral oil),在转化时如果把矿物油带入感受态菌,会严重影响转化效率。
- 感受态细菌效率不够高,请检测一下感受态细胞的效率,确保转化效率在107以上,当然高一些更好。
- 有克隆,但没有或很难检测到预期的突变克隆:
- DpnI消化效果不佳。一种可能是加入DpnI后,由于该酶在甘油中,会迅速下沉,如果没有混匀就会严重影响DpnI的消化作用。另一种可能是DpnI由于保存不当或保存时间过长等原因导致活性下降,这时可以适当延长消化的时间至1~2小时。
- 使用的待突变的模板质粒量过多,导致DpnI消化时不完全。我们这里推荐的模板质粒用量0.5μg,已经是模板质粒用量的上限,不能再使用更多的模板质粒了。
- 尽量避免多次反复冻融dNTP。可以把dNTP适当分装后再使用。
- DpnI消化效果不佳。一种可能是加入DpnI后,由于该酶在甘油中,会迅速下沉,如果没有混匀就会严重影响DpnI的消化作用。另一种可能是DpnI由于保存不当或保存时间过长等原因导致活性下降,这时可以适当延长消化的时间至1~2小时。
- 有突变克隆,但突变位点不是预期的位点:
- 引物设计不佳,并且PCR反应中退火温度过低,导致引物退火到错误的地方。
- 引物质量较差,没有经过PAGE纯化。这样引物中通常会含有比设计的引物要短的特异性较差的引物,容易导致非预期的突变。
- 引物设计不佳,并且PCR反应中退火温度过低,导致引物退火到错误的地方。
相关搜索:基因定点突变试剂盒,定点突变试剂盒,Site-directed Gene Mutagenesis Kit